MetaGraph
MetaGraph is a C++ framework library for indexing very large collections of DNA/Protein sequences and a tool for scalable construction of annotated genome graphs and sequence-to-graph alignment.
Although the target use cases of MetaGraph overlap with BLAST, MetaGraph mainly focuses on the scalable indexing of raw sequencing data in annotated de Bruijn graphs with up to $\sim 10^{12}$ nodes and $\sim 10^{7}$ annotation labels. It also provides an online platform MetaGraph Online.
The default index representations in MetaGraph are extremely scalable and support building graphs with trillions of nodes and millions of annotation labels. At the same time, the provided workflows and their careful implementation, combined with low-level optimizations of the core data structures, enable exceptional query and alignment performance.
Main features:
- Large-scale indexing of sequences
- Python API for querying in the server mode
- Support for representing k-mer counts or expression values
- Sequence alignment against very large annotated graphs
- Scalable cleaning of very large de Bruijn graphs (to remove sequencing errors)
- Support for custom alphabets (e.g., {A,C,G,T,N} or amino acids)
- Algorithms for differential assembly
Design choices in MetaGraph:
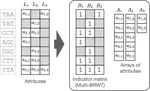
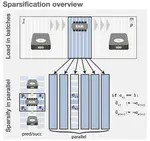
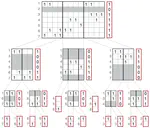
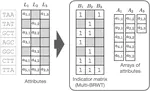
- Use of succinct data structures and efficient representation schemes for extremely high scalability
- Algorithmic choices that work efficiently with succinct data structures (e.g., always prefer batched operations)
- Modular support of different graph and annotation representations
- Use of generic and extensible interfaces to support adding custom index representations / algorithms with little code overhead.
Other contributors: Marc Zimmermann, Thomas Zhou, the MetaGraph team.
Mikhail Karasikov
ML Engineer, PhD
Machine learning researcher/engineer at kaiko.ai with a background in mathematics and computer science.